Press Releases Archive
07.02.2018
Alzheimer’s research on mice – intracellular calcium store malfunction leads to brain hyperactivity
Physiologists at the University of Tübingen discover the mechanism leading to increased release of neurotransmitters – and potential new approaches to treatment
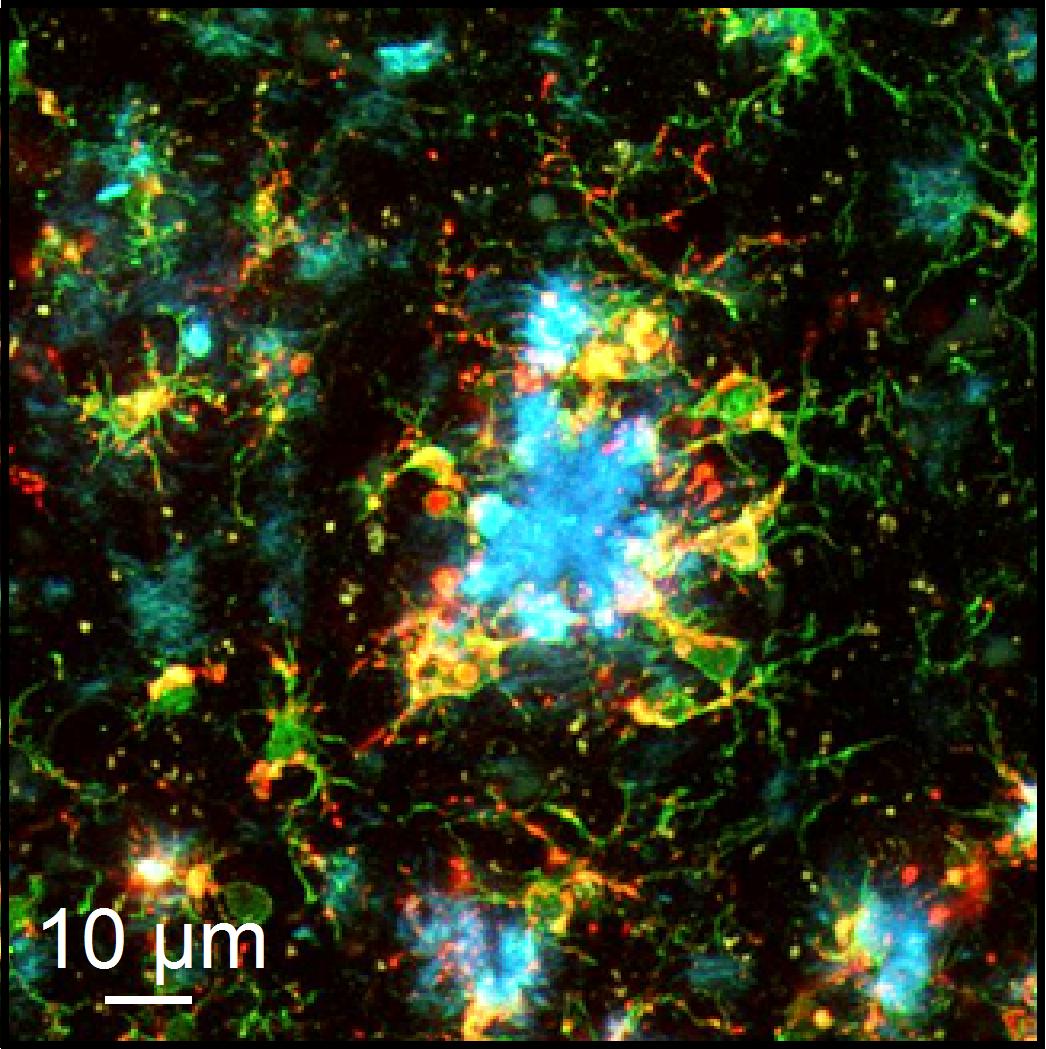
Alzheimer´s disease is the key cause of dementia in elderly patients. Those affected develop deficiencies in their abilities to learn, think logically, communicate, and to master the challenges of everyday life. To find out more about how the disease comes about, researchers at the University of Tübingen used mice, developing the same amyloid protein deposits in their brains (see illustration) as human patients, and which also suffer memory loss. Several years ago, a team led by Professor Olga Garaschuk showed that in these mice, the disease coincided with a noticeable increase in nerve cell activity in the brain. There were similar findings in human Alzheimer’s patients. Now, Garaschuk’s team at the University of Tübingen’s Institute of Physiology can explain an important mechanism behind this neural hyperactivity in mice. At the contact points between nerve cells, there is a malfunction in the intracellular calcium storage which is needed for signal transfer. As a result, too many signal chemicals (neurotransmitters) are released into the synaptic cleft. The study, published in the latest edition of PNAS, shows how new findings can lead to fresh approaches to treatment of the hereditary form of Alzheimer´s disease.
Communication between nerve cells in the brain is largely carried out via electrical signals. But at the synapse – the transfer point between one nerve cell and another – the signal switches to a chemical one. Calcium plays an important role here; it helps to release messenger chemicals known as neurotransmitters. They dock onto the next nerve cell, where another electrical impulse is generated and sent on. In the new study, Garaschuk found that in mouse models of Alzheimer’s disease showing this abnormal increase in brain nerve cell activity, calcium storage at the presynaptic side was dysregulated. “This releases a larger amount of neurotransmitters into the cerebral cortex – which leads to hyperactivity in the nerve cells,” she explains.
Alzheimer’s occurs sporadically in humans, the greatest risk factor being age. Yet a proportion of Alzheimer’s patients also have a genetic tendency towards the disease. In this kind of Alzheimer´s, 90 percent of those affected have a mutation of the presenilin gene. “Interestingly, in mice, one single copy of this kind of mutated gene is enough to cause hyperactivity due to calcium storage malfunction,” Garaschuk says. Chemical agents which can empty the cell’s calcium storage or – as one clinically approved drug does – block the release of calcium from this store; they also suppress the cell hyperactivity. “That leads to a normalisation of cerebral cortex functions,” she says. These findings could play a role in the development of new treatments for Alzheimer’s disease.
Publication:
Chommanad Lerdkrai, Nithi Asavapanumas, Bianca Brawek, Yury Kovalchuk, Nima Mojtahedi, Maria Olmedillas del Moral, and Olga Garaschuk: Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. PNAS, DOI 10.1073/pnas.1714409115
Contact:
Professor Dr. Olga Garaschuk
University of Tübingen
Faculty of Medicine
Institute of Physiology
Phone +49 7071 29-73640
olga.garaschuk@uni-tuebingen.de